AnnData/Seurat Import
spacedeconv_import.RmdSetup
The preferred single-cell reference data input format of
spacedeconv is SingleCellExperiment.
Other common single-cell formats are anndata and
Seurat. This vignette shows how to use
H5AD anndata files and
Seurat objects as input for the spacedeconv pipeline.
For anndata, we use the
zellkonverter package which is also available via
conda.
Here, we set up reticulate before loading the
spacedeconv and zellkonverter libraries
to avoid reticulate conflicts between the two
packages.
library(reticulate)
use_condaenv("spacedeconv-env", required = TRUE)
invisible(py_config())
library(spacedeconv)
library(zellkonverter)
library(Seurat)
library(SingleCellExperiment)
SingleCellExperiment <-> AnnData Conversion
zellkonverter allows us to convert
SingleCellExperiment objects to
anndata and store them as H5AD files:
zellkonverter::writeH5AD()
writeH5AD(single_cell_data_3, "single_cell_data_3.h5ad")
In case we have the single-cell data only as H5AD
anndata file, we can use
zellkonverter to read it into R as
SingleCellExperiment object:
zellkonverter::readH5AD()
That way it can be used as input for the
spacedeconv pipeline. In this case we read back the
example data file we just wrote to H5AD.
single_cell_data_3 <- readH5AD("single_cell_data_3.h5ad", use_hdf5 = FALSE)
SingleCellExperiment <-> Seurat Conversion
Seurat allows us to convert
SingleCellExperiment to Seurat objects:
Seurat::as.Seurat()
single_cell_data_3 <- as.Seurat(single_cell_data_3, counts = "counts", data = NULL)
If you have single-cell data as Seurat object, you
can convert it to a SingleCellExperiment to use it as
input for spacedeconv:
Seurat::as.SingleCellExperiment()
single_cell_data_3 <- as.SingleCellExperiment(single_cell_data_3, assay = "originalexp")
Spacedeconv Clustering Pipeline
Now we can continue with the spacedeconv pipeline as usual. In this case we will perform the clustering procedure as an example.
single_cell_data_3 <- spacedeconv::preprocess(single_cell_data_3)
spatial_data_3 <- spacedeconv::preprocess(spatial_data_3)
spatial_data_3 <- spacedeconv::normalize(spatial_data_3, method = "cpm")
signature <- spacedeconv::build_model(
single_cell_obj = single_cell_data_3,
cell_type_col = "celltype_major",
method = "spatialdwls",
verbose = TRUE
)
#> class of selected matrix: dgCMatrix
#> [1] "finished runPCA_factominer, method == factominer"
deconv <- spacedeconv::deconvolute(
spatial_obj = spatial_data_3,
single_cell_obj = single_cell_data_3,
cell_type_col = "celltype_major",
method = "spatialdwls",
signature = signature,
assay_sp = "cpm"
)
#> class of selected matrix: dgCMatrix
#> [1] "finished runPCA_factominer, method == factominer"
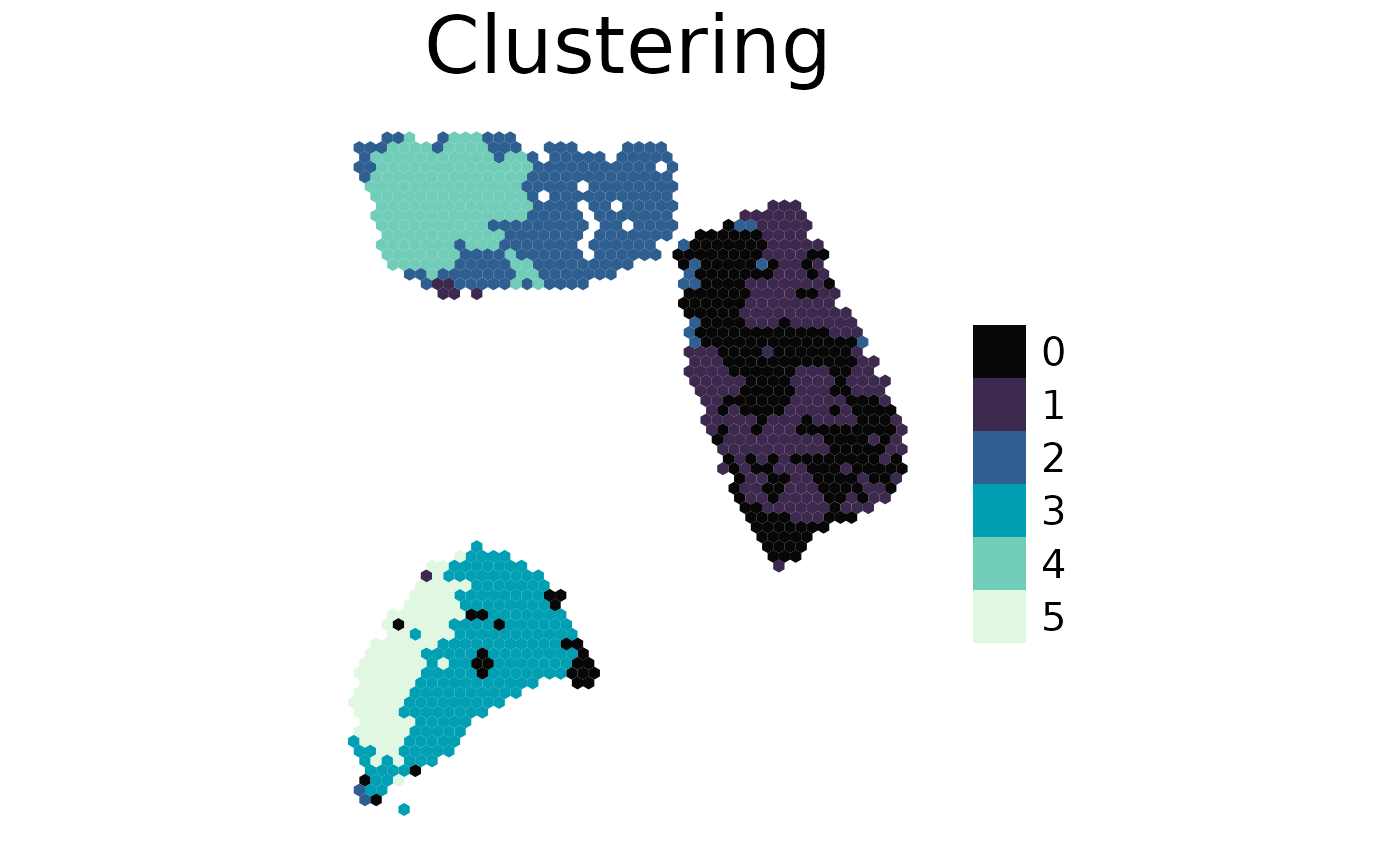
clustered <- spacedeconv::cluster(deconv, data = "expression", clusres = 0.5)
spacedeconv::plot_spatial(
spe = clustered,
result = "cluster_expression_res_0.5",
title = "Clustering",
density = FALSE
)