![]()
spacedeconv is a unified interface to first- and second-generation deconvolution tools with focus on spatial transcriptomics datasets. The package is able to directly estimate celltype proportions of immune cells and can deconvolute any celltype if an annotated single-cell reference dataset is available.
⬇️ Installation
Note: The current spacedeconv installation is only available for the linux-64 platform.
Since many different packages need to be included, we highly recommend to install spacedeconv in a new Conda environment with the following commands.
First, a tool for fast dependency resolution is needed, therefore we recommend installing micromamba (or mamba / conda with libmamba solver) if not already available:
Download the environment.yml file of this github repo:
wget https://raw.githubusercontent.com/omnideconv/spacedeconv/main/environment.yml -O environment.ymlCreate a new environment called “spacedeconv-env” via micromamba with the environment.yml file and flexible channel priority:
Start R inside the spacedeconv-env conda environment:
Install the missing packages that are not available via conda as well as spacedeconv:
pak::pkg_install("drieslab/Giotto@v3.3.2", dependencies = FALSE, upgrade = FALSE)
pak::pkg_install("YingMa0107/CARD@2d64b91abb5cdd0c7f576b1c5d4727c84e7c93a0", dependencies = FALSE, upgrade = FALSE)
pak::pkg_install("omnideconv/spacedeconv", dependencies = FALSE, upgrade = FALSE)
✨ Features
- unified access to first- and second-generation deconvolution tools
- direct deconvolution of immune cells
- compute custom reference signatures to deconvolute any celltype
- flexible visualization functions
- resource optimization
- Pathway and Transcription Factor analysis (decoupleR integration)
- Ligand-Receptor analysis
- easy integration into spatial transcriptomics workflows
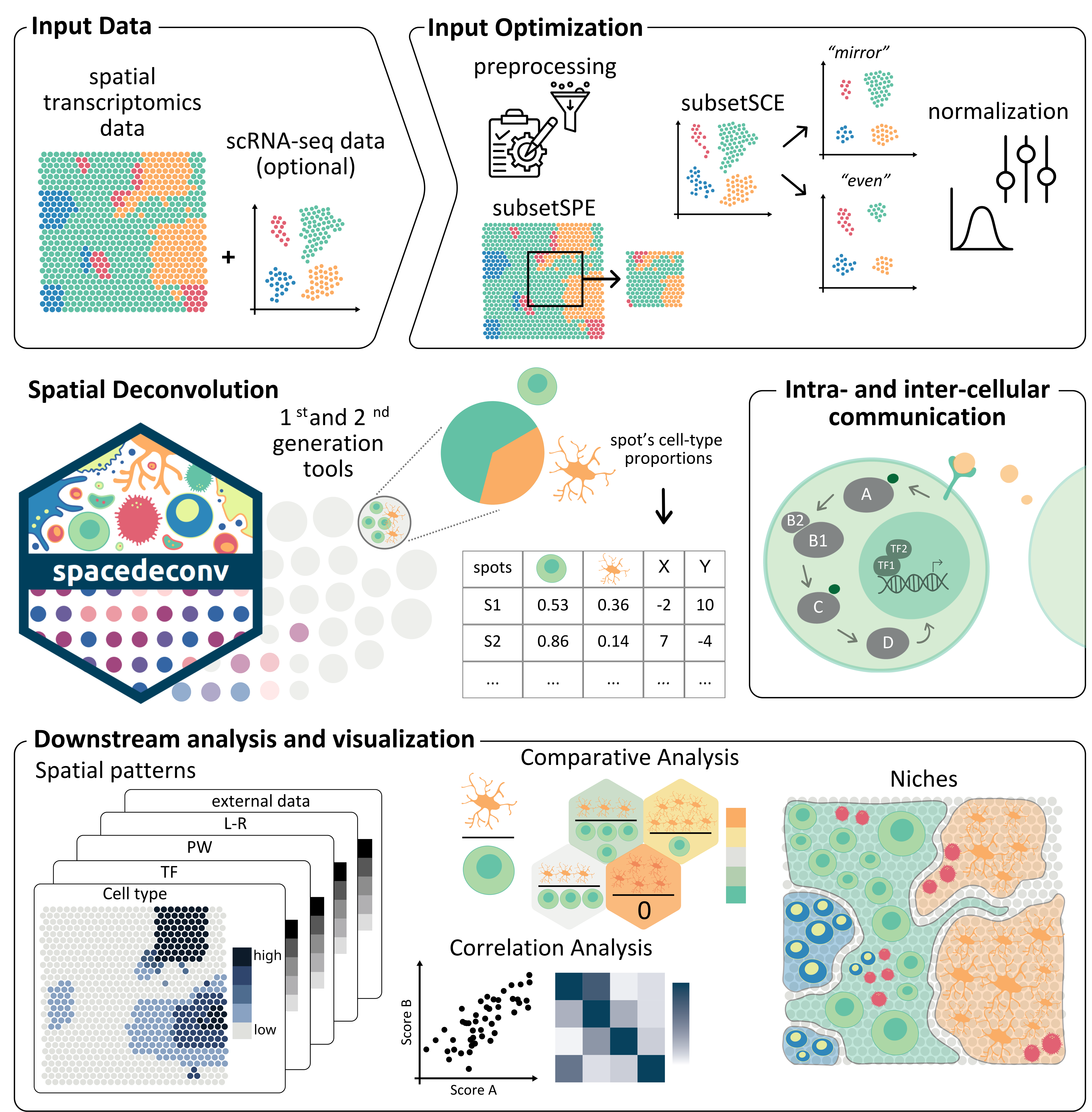
💾 Data requirements
Spatial transcriptomics data: SpatialExperiment
Single-cell data with cell-type annotation: SingleCellExperiment (recommended) or Seurat
🧑💻 Usage
The main workflow consists of the following steps:
1. Load datasets
To explore the package, start by loading some of the built-in example datasets.
library(spacedeconv)
# data("single_cell_data_1")
# data("single_cell_data_2")
data("single_cell_data_3")
# data("single_cell_data_4")
# data("spatial_data_1")
# data("spatial_data_2")
data("spatial_data_3")
# data("spatial_data_4")
2. Preprocessing
Depending on the use case, certain preprocessing steps might be necessary.
single_cell_data_3 <- spacedeconv::preprocess(single_cell_data_3)
spatial_data_3 <- spacedeconv::preprocess(spatial_data_3)
spatial_data_3 <- spacedeconv::normalize(spatial_data_3, method = "cpm")
3. Build a Signature Matrix
Build a cell type specific signature matrix from annotated single-cell reference data.
signature <- spacedeconv::build_model(
single_cell_obj = single_cell_data_3,
cell_type_col = "celltype_major",
method = "spatialdwls", verbose = T
)
You can see all supported deconvolution methods with
spacedeconv::deconvolution_methods.
4. Deconvolution
While some methods are able to directly estimate immune cell abundances other tools require a custom reference signature.
deconv <- spacedeconv::deconvolute(
spatial_obj = spatial_data_3,
single_cell_obj = single_cell_data_3,
cell_type_col = "celltype_major",
method = "spatialdwls",
signature = signature,
assay_sp = "cpm"
)
5. Visualization
spacedeconv includes highly-flexible visualization functions.
plot_spatial(
spe = deconv,
result = "spatialdwls_B.cells",
title = "B cells",
density=F
)
Available methods, Licenses, Citations
Note that, while spacedeconv itself is free (GPL 3.0), you may need to obtain a license to use the individual methods. See the table below for more information. If you use this package in your work, please cite both our package and the method(s) you are using.
Constantin Zackl, Maria Zopoglou, Reto Stauffer, Markus Ausserhofer, Marieke E. Ijsselsteijn, Gregor Sturm, Noel Filipe da Cunha Carvalho de Miranda, Francesca Finotello. spacedeconv: deconvolution of tissue architecture from spatial transcriptomics, PREPRINT available at Research Square https://doi.org/10.21203/rs.3.rs-5102166/v1.
| Method | Usable with signature() | Licence | Citation |
|---|---|---|---|
| immunedeconv | ❌ | BSD | Sturm, G. et al. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics, 35(14), i436-i445 (2019). https://doi.org/10.1093/bioinformatics/btz363 |
| spatialDWLS | ✔️ | MIT | Dong R., Yuan G.C. SpatialDWLS: accurate deconvolution of spatial transcriptomic data. Genome Biology 22, 145 (2021). https://doi.org/10.1186/s13059-021-02362-7 |
| cell2location | ✔️ | Apache-2.0 | Kleshchevnikov V. et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nature Biotechnology 40, 661–671 (2022). https://doi.org/10.1038/s41587-021-01139-4 |
| SPOTlight | ✔️ | GPL 3.0 | Elosua-Bayes M. et al. SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Research 49(9):e50 (2021). https://doi.org/10.1093/nar/gkab043. |
| RCTD | ✔️ | GPL 3.0 | Cable D.M. et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nature Biotechnology 40, 517–526 (2022). https://doi.org/10.1038/s41587-021-00830-w |
| CARD | ✔️ | GPL-3.0 | Ma Y., Zhou X. Spatially informed cell type deconvolution for spatial transcriptomics. Nature Biotechnology 40, 1349–1359 (2022). https://doi.org/10.1038/s41587-022-01273-7 |
| DOT | ✔️ | GPL-3.0 | Rahimi A. et al. DOT: a flexible multi-objective optimization framework for transferring features across single-cell and spatial omics. Nature Communications 15, 4994 (2024). https://www.nature.com/articles/s41467-024-48868-z |