Ligand-Receptor Analysis
2024-02-27
spacedeconv_LR.Rmd
library(spacedeconv)
#> → Setting up spacedeconv environment..
#> → Using conda environment 'spacedeconv-env'
library(OmnipathR)
library(SpatialExperiment)
#> Loading required package: SingleCellExperiment
#> Loading required package: SummarizedExperiment
#> Loading required package: MatrixGenerics
#> Loading required package: matrixStats
#>
#> Attaching package: 'MatrixGenerics'
#> The following objects are masked from 'package:matrixStats':
#>
#> colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
#> colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
#> colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
#> colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
#> colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
#> colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
#> colWeightedMeans, colWeightedMedians, colWeightedSds,
#> colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
#> rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
#> rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
#> rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
#> rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
#> rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
#> rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
#> rowWeightedSds, rowWeightedVars
#> Loading required package: GenomicRanges
#> Loading required package: stats4
#> Loading required package: BiocGenerics
#>
#> Attaching package: 'BiocGenerics'
#> The following object is masked from 'package:spacedeconv':
#>
#> normalize
#> The following objects are masked from 'package:stats':
#>
#> IQR, mad, sd, var, xtabs
#> The following objects are masked from 'package:base':
#>
#> anyDuplicated, aperm, append, as.data.frame, basename, cbind,
#> colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
#> get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply,
#> match, mget, order, paste, pmax, pmax.int, pmin, pmin.int,
#> Position, rank, rbind, Reduce, rownames, sapply, setdiff, sort,
#> table, tapply, union, unique, unsplit, which.max, which.min
#> Loading required package: S4Vectors
#>
#> Attaching package: 'S4Vectors'
#> The following object is masked from 'package:utils':
#>
#> findMatches
#> The following objects are masked from 'package:base':
#>
#> expand.grid, I, unname
#> Loading required package: IRanges
#> Loading required package: GenomeInfoDb
#> Loading required package: Biobase
#> Welcome to Bioconductor
#>
#> Vignettes contain introductory material; view with
#> 'browseVignettes()'. To cite Bioconductor, see
#> 'citation("Biobase")', and for packages 'citation("pkgname")'.
#>
#> Attaching package: 'Biobase'
#> The following object is masked from 'package:MatrixGenerics':
#>
#> rowMedians
#> The following objects are masked from 'package:matrixStats':
#>
#> anyMissing, rowMedians
Introduction
This function provides a framework to infer Ligand-Receptor Interactions within a sample using known L-R pairs. The function extracts the expression levels of known L-R pairs from the gene expression matrix of a given spe object.
Example Data
We use spacedeconvs spatial data 3 for this analysis. We always encourage the user to preprocess and normalize the data first, but in this case, the function would also preprocess and normalize the data to CPM internally, so the following step is optional.
data("spatial_data_3")
# this step is optional
spe <- spacedeconv::preprocess(spatial_data_3, remove_mito = TRUE)
spe <- spacedeconv::normalize(spe, method = "cpm")
L-R Analysis
The user can define a reference table with L-R pairs or use the default table, which is a filtered selection of the intercell network, as provided by Omnipath (the user can either leave the resource argument empty or input “Consensus” to use the default resource). In the case of a user-defined L-R table, this should contain the following columns: source_symbol column containing the ligands or ligand complexes and the target_symbol column with the receptors or receptor complexes.
Complexes: The function can also handle L or R complexes. In this case, the members of the complex are split and their expressions are extracted separately. If one of the members of either the Ligand- or the Receptor- complex is missing from the CPM expression matrix of the spe object then the L-R pair is removed.
Calculation of scores L-R scores can be calculated using two mathematical approaches, which can be defined using the method argument. Either the min values between Ligand and Receptors in each pair (deafult or input “min”) or the product of the expression of the Ligand and the Receptor for each pair (input “product”)
Organisms: The function can also handle mouse data. The user can input a user-defined reference table with L–R mouse pairs. The user can convert human to mouse L-R pairs with the convert_mouse_to_human function of spacedeconv. The user-defined table should have the same format as that mentioned above. Otherwise the default human reference table is converted internally to mouse data and used to calculate the L-R scores. To use the default mouse table input “mouse” in the organism argument and either leave the resource parameter empty or input “Consensus”
After the calculation the scores for each L-R pair are stored in the ColData of the spe object and are marked with a “lr”.
The example Data used here, is a human breast cancer slide and we will use the “min” mathematical approach with the default reference L-R table (“Consensus”).
spe <- spacedeconv::get_lr(spe,
resource = "Consensus",
method = "min",
organism = "human"
)
Visualization of the results.
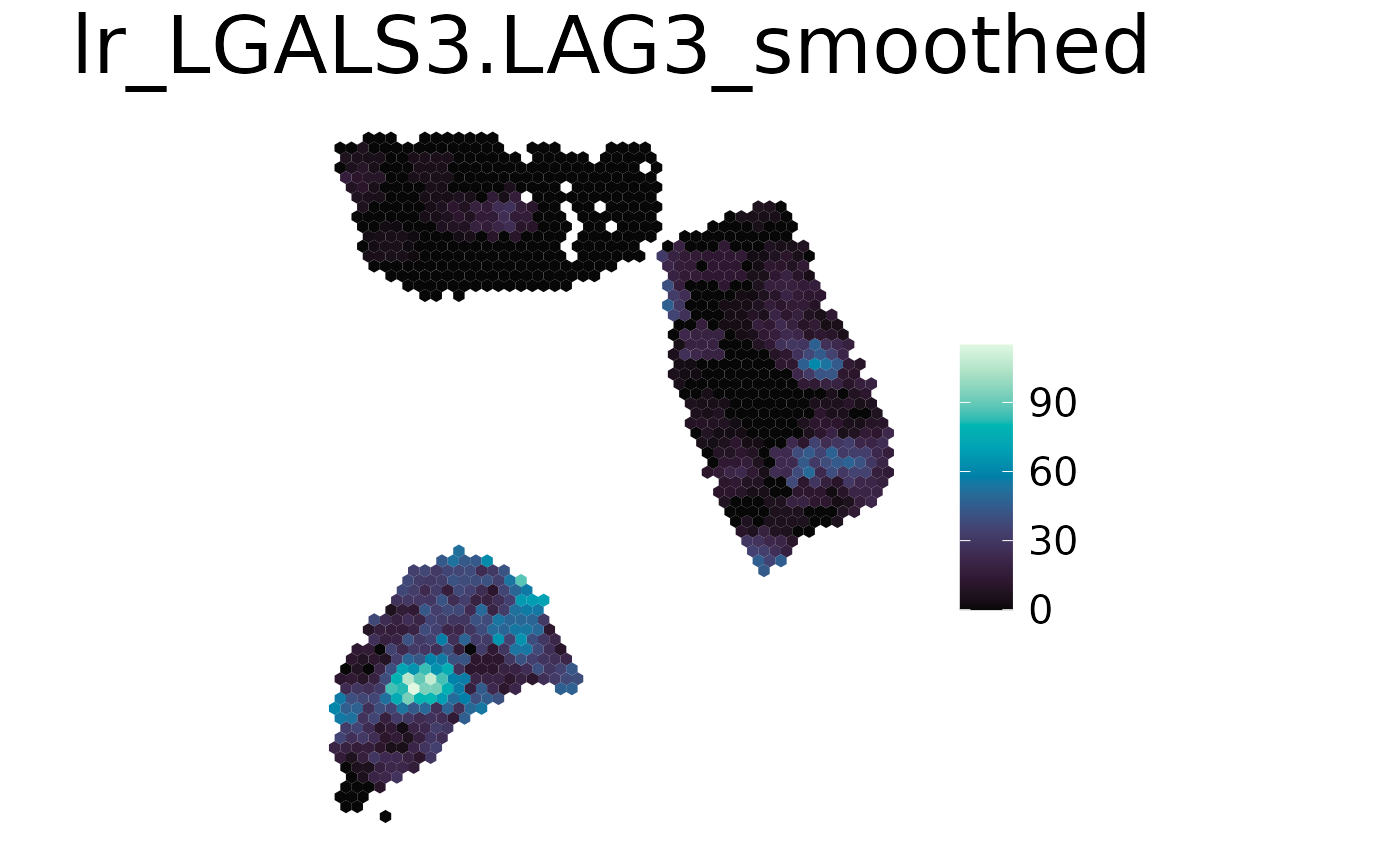
The results can be visualized with the plot_celltype function of spacedeconv.
spacedeconv::plot_spatial(spe,
result = "lr_LGALS3.LAG3",
smooth = TRUE,
density = FALSE
)
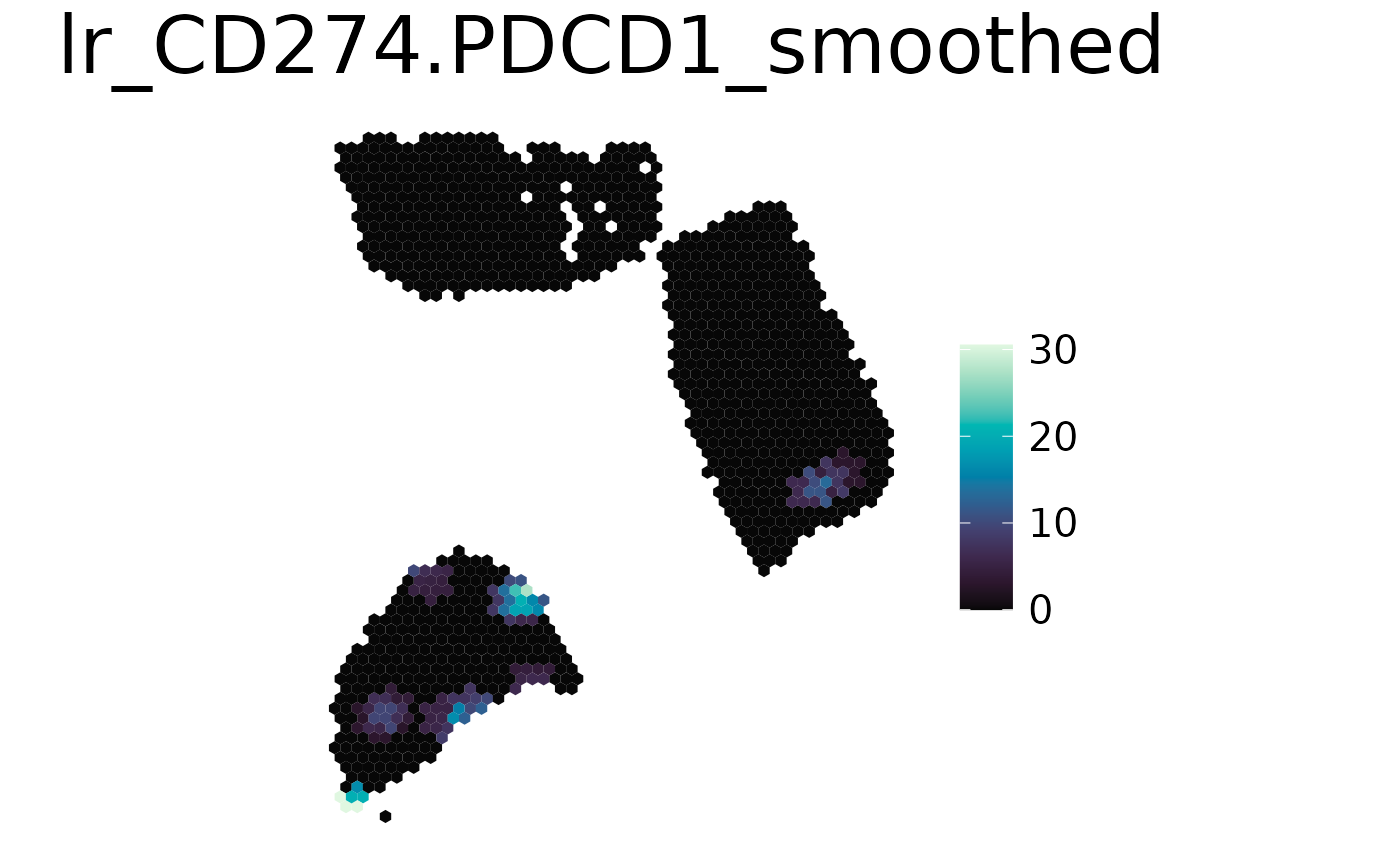
spacedeconv::plot_spatial(spe,
result = "lr_CD274.PDCD1",
smooth = TRUE,
density = FALSE
)