Deconvolution of the tumor microenvironment with omnideconv
Source:vignettes/omnideconv_example.Rmd
omnideconv_example.Rmd1. Introduction and dataset
In this vignette, we will use the omnideconv package to deconvolve a
bulk RNA-seq dataset from 24 breast cancer patients with two different
methods (DWLS and BayesPrism). The datasets are from a recent breast
cancer study (Wu et al.
2021). This study provides access to a primary-tumor single
cell RNA-seq (scRNA-seq) dataset from 26 breast cancer patients across
three major cancer subtypes (ER+, HER2+, TNBC). The dataset includes
cell-type annotation for three resolution levels. In addition, the data
includes bulk RNA-seq sequencing for 24 of the patients. In this
chapter, we use multi-sample, breast-cancer scRNA-seq atlas (100,064
cells) as a reference to train the methods for the deconvolution of the
bulk RNA-seq samples. The single-cell and bulk RNA-seq data is deposited
on GEO, under accession number: GSE176078.
The single cell data comes with the author’s cell type annotations. We
will need to download and unzip the datasets
(GSE176078_Wu_etal_2021_BRCA_scRNASeq.tar.gz,
GSE176078_Wu_etal_2021_bulkRNAseq_raw_counts.txt.gz), and
store them in a data directory. For this example analysis, we will also
need the additional clinical information about the patients, available
as Supplementary
Table 1 from the paper.
3. Single cell data processing
Although not strictly required by omnideconv, we suggest performing quality control and filtering of the input scRNA-seq data according to the best practice (Heumos et al. 2023), to ensure the best training conditions for deconvolution algorithms. We first pre-process the single-cell dataset to remove low-quality cells. We will use the R package Seurat (Hao et al. 2023), which allows fast and easy manipulation of single-cell data. We will create a Seurat object with the cell counts and their metadata of interest curated by the authors, which include cell-type annotation on three levels of resolution:
single.cell.data <- Seurat::ReadMtx(
mtx = "path/to/Wu_etal_2021_BRCA_scRNASeq/count_matrix_sparse.mtx",
cells = "path/to/Wu_etal_2021_BRCA_scRNASeq/count_matrix_barcodes.tsv",
features = "path/to/Wu_etal_2021_BRCA_scRNASeq/count_matrix_genes.tsv",
feature.column = 1
)
single.cell.metadata <- read.table("path/to/Wu_etal_2021_BRCA_scRNASeq/metadata.csv",
sep = ",", header = TRUE, row.names = 1
)
single.cell.data <- CreateSeuratObject(single.cell.data,
project = "Wu_dataset", assay = "RNA",
min.cells = 0, min.features = 1,
meta.data = single.cell.metadata
)We can have an overview of the number of cells per cell type in the dataset:
| celltype_major | number_cells |
|---|---|
| B-cells | 3206 |
| CAFs | 6573 |
| Cancer Epithelial | 24489 |
| Endothelial | 7605 |
| Myeloid | 9675 |
| Normal Epithelial | 4355 |
| Plasmablasts | 3524 |
| PVL | 5423 |
| T-cells | 35214 |
In order to remove low quality cells, we will follow the best practices for single cell normalization (Heumos et al. 2023). We will perform quality control on each cell by considering metrics such as the number of total counts, the number of expressed features (genes), and the fraction of mitochondrial genes per cell. We will remove cells that have , where
- is the respective metric of an observation (cell), for example the number of detected genes
- is the Median Absolute Deviation, computed as
- is set to 5 for the number of counts and expressed features, and to 3 for the fraction of mitochondrial genes
In order to do this filtering, we will create a function to identify the outliers for each metric:
is_outlier <- function(SeuratObject, metric, nmads) {
eval(parse(text = paste0("M <- SeuratObject$", metric)))
outlier <- (M < median(M) - nmads * mad(M)) | (M > median(M) + nmads * mad(M))
return(outlier)
}
check_outliers_nFeature <- is_outlier(single.cell.data, "nFeature_RNA", 5)
check_outliers_nCount <- is_outlier(single.cell.data, "nCount_RNA", 5)
check_outliers_mito <- is_outlier(single.cell.data, "percent.mito", 3)
non_outliers_nFeature <- names(check_outliers_nFeature[!check_outliers_nFeature])
non_outliers_nCount <- names(check_outliers_nCount[!check_outliers_nCount])
non_outliers_mito <- names(check_outliers_mito[!check_outliers_mito])
non_outliers <- intersect(non_outliers_nFeature, non_outliers_nCount) %>%
intersect(non_outliers_mito)
single.cell.data <- subset(single.cell.data, cells = non_outliers)| celltype_major | number_cells |
|---|---|
| B-cells | 3150 |
| CAFs | 6154 |
| Cancer Epithelial | 16613 |
| Endothelial | 6991 |
| Myeloid | 8855 |
| Normal Epithelial | 3533 |
| Plasmablasts | 3164 |
| PVL | 5120 |
| T-cells | 34991 |
4. Bulk data preprocessing
We will now read in the bulk sequencing data file, which consists of 24 samples.
bulk.data <- read.table("path/to/GSE176078_Wu_etal_2021_bulkRNAseq_raw_counts.txt", skip = 1)
header <- read.table("path/to/GSE176078_Wu_etal_2021_bulkRNAseq_raw_counts.txt",
header = FALSE, nrows = 1, skipNul = TRUE, sep = "\t"
)
colnames(bulk.data) <- c("Genes", gsub("A|N", "", header[2:25]))
bulk.data <- bulk.data[bulk.data$Genes != "", ]
bulk.data <- column_to_rownames(bulk.data, "Genes")
bulk.data <- as.matrix(bulk.data)5. Subsampling of single cell data
The various methods included in omnideconv rely on the single cell dataset that will be used to train them for the deconvolution of those specific cell types. This training involves the optimization of internal features of the methods and can happen in different ways. Some methods use the single cell data to build a ‘signature matrix’, i.e. a reduced transcriptional fingerprints of the cell types provided, while others use this data in a statistical or deep learning model. Since single cell datasets can often encompass thousands of cells, we will need to subsample it in order to be able to run the analysis on our machines. In this case we will retain a maximum of 200 cells per cell type, but this step can be customized, or eventually skipped, depending on the computational resources available.
max_cells_per_celltype <- 200
sampled.metadata <- single.cell.data@meta.data %>%
rownames_to_column("barcode") %>%
group_by(celltype_major) %>%
nest() %>%
mutate(n = map_dbl(data, nrow)) %>%
mutate(n = min(n, max_cells_per_celltype)) %>%
ungroup() %>%
mutate(samp = map2(data, n, sample_n)) %>%
select(-data) %>%
unnest(samp)
single.cell.data.sampled <- subset(single.cell.data, cells = sampled.metadata$barcode)| celltype_major | number_cells |
|---|---|
| B-cells | 200 |
| CAFs | 200 |
| Cancer Epithelial | 200 |
| Endothelial | 200 |
| Myeloid | 200 |
| Normal Epithelial | 200 |
| Plasmablasts | 200 |
| PVL | 200 |
| T-cells | 200 |
6. Deconvolution of the bulk data
Each method has different requirements, but in general to compute the deconvolution results we will need the single cell counts matrix, the cell type annotations and the information on the individual/experiment from which the cells were retrieved (batch ID).
counts.matrix <- as.matrix(single.cell.data.sampled@assays$RNA@counts)
cell.type.annotations <- single.cell.data.sampled$celltype_major
batch.ids <- single.cell.data.sampled$orig.ident6.1 Deconvolution with DWLS
Now we’re going to deconvolute the bulk dataset with different
methods. The first one we are going to use is called DWLS (Tsoucas et al.
2019) and performs the deconvolution in a two-step process.
First, the single cell data is used to build a signature matrix using
the omnideconv function build_model. DWLS
looks for differentially expressed genes that discriminate across cell
types, and can do so with two approaches based either on the Seurat
(Hao et al.
2023) “bimod” test (McDavid et al. 2012) or on MAST (Finak et al.
2015). MAST improves the former model, but has an increased
computational requirement (Nault et al. 2022). The authors
recommend using this method on the smaller datasets, and to switch to
Seurat if the analysis with MAST cannot be completed. To reduce MAST’s
computational time, we introduced a second version of the MAST-based
function (mast_optimized) that speeds up the process compared to the
original implementation:
signature.matrix.dwls <- omnideconv::build_model(
single_cell_object = counts.matrix,
cell_type_annotations = cell.type.annotations,
method = "dwls",
dwls_method = "mast_optimized"
)This signature is optimized so that the genes selected are the ones that help to discriminate across cell types.
The signature is now used for the deconvolution of the bulk RNAseq,
which is performed with the omnideconv function
deconvolute. DWLS computes the cell fractions performing
one of Ordinary Least Squares (OLS) Regression, Support Vector
Regression (SVR) or the Dampened Weighted Least Squares Regression
(DWLS) that was introduced with the method. This last regression method
is shown to outperform the others when it comes to the detection of rare
cell types:
deconvolution.results.dwls <- deconvolute(
bulk_gene_expression = bulk.data,
model = signature.matrix.dwls,
method = "dwls",
dwls_submethod = "DampenedWLS"
)We will now obtain, for every sample, a set of cell type fractions for each cell type that was included in the provided single cell dataset.
| B-cells | CAFs | Cancer Epithelial | Endothelial | Myeloid | Normal Epithelial | Plasmablasts | PVL | T-cells | |
|---|---|---|---|---|---|---|---|---|---|
| CID3586 | 0.231 | 0.238 | 0.170 | 0.083 | 0.005 | 0.035 | 0.000 | 0.022 | 0.215 |
| CID3921 | 0.026 | 0.177 | 0.145 | 0.114 | 0.019 | 0.068 | 0.004 | 0.000 | 0.447 |
| CID3941 | 0.000 | 0.267 | 0.373 | 0.091 | 0.002 | 0.180 | 0.000 | 0.030 | 0.059 |
| CID3948 | 0.466 | 0.107 | 0.264 | 0.049 | 0.007 | 0.000 | 0.001 | 0.020 | 0.086 |
| CID3963 | 0.112 | 0.208 | 0.000 | 0.088 | 0.103 | 0.345 | 0.000 | 0.020 | 0.123 |
| CID4066 | 0.049 | 0.342 | 0.174 | 0.102 | 0.019 | 0.155 | 0.000 | 0.049 | 0.111 |
| CID4067 | 0.000 | 0.338 | 0.477 | 0.084 | 0.007 | 0.018 | 0.000 | 0.006 | 0.071 |
| CID4290 | 0.000 | 0.408 | 0.417 | 0.068 | 0.072 | 0.005 | 0.000 | 0.024 | 0.006 |
| CID4398 | 0.000 | 0.220 | 0.143 | 0.119 | 0.006 | 0.204 | 0.000 | 0.034 | 0.275 |
| CID44041 | 0.332 | 0.161 | 0.000 | 0.122 | 0.006 | 0.264 | 0.000 | 0.032 | 0.084 |
| CID4461 | 0.319 | 0.138 | 0.292 | 0.084 | 0.028 | 0.000 | 0.003 | 0.020 | 0.116 |
| CID4463 | 0.033 | 0.123 | 0.560 | 0.069 | 0.067 | 0.036 | 0.000 | 0.039 | 0.074 |
| CID4465 | 0.226 | 0.287 | 0.066 | 0.099 | 0.070 | 0.206 | 0.004 | 0.013 | 0.029 |
| CID4471 | 0.009 | 0.288 | 0.000 | 0.182 | 0.046 | 0.296 | 0.000 | 0.143 | 0.035 |
| CID4495 | 0.366 | 0.066 | 0.023 | 0.071 | 0.066 | 0.274 | 0.002 | 0.005 | 0.126 |
| CID44971 | 0.286 | 0.099 | 0.000 | 0.083 | 0.002 | 0.300 | 0.000 | 0.010 | 0.219 |
| CID4513 | 0.001 | 0.398 | 0.000 | 0.143 | 0.397 | 0.000 | 0.001 | 0.056 | 0.005 |
| CID4515 | 0.456 | 0.185 | 0.000 | 0.092 | 0.050 | 0.131 | 0.002 | 0.000 | 0.085 |
| CID4523 | 0.000 | 0.219 | 0.381 | 0.165 | 0.025 | 0.166 | 0.000 | 0.000 | 0.044 |
| CID4530 | 0.085 | 0.317 | 0.284 | 0.083 | 0.034 | 0.068 | 0.000 | 0.040 | 0.088 |
| CID4535 | 0.075 | 0.062 | 0.681 | 0.049 | 0.007 | 0.000 | 0.000 | 0.029 | 0.097 |
| CID4040 | 0.305 | 0.217 | 0.272 | 0.052 | 0.030 | 0.000 | 0.002 | 0.009 | 0.114 |
| CID3838 | 0.169 | 0.200 | 0.332 | 0.085 | 0.058 | 0.077 | 0.000 | 0.027 | 0.052 |
| CID3946 | 0.389 | 0.147 | 0.000 | 0.081 | 0.038 | 0.256 | 0.000 | 0.004 | 0.085 |
We can also visualise the results as a barplot through the built-in
plot_deconvolution function:
omnideconv::plot_deconvolution(list("dwls" = deconvolution.results.dwls), "bar", "method", "Spectral")6.2 Deconvolution with BayesPrism
The third method we will use is BayesPrism (Chu et al. 2022). This method is based on a Bayesian framework and models the transcriptomic expression observed in the scRNA-seq dataset. It then uses this information to dissect the bulk RNA-seq.
deconvolution.results.bayesprism <- deconvolute(
bulk_gene_expression = bulk.data,
single_cell_object = counts.matrix,
cell_type_annotations = cell.type.annotations,
model = NULL,
method = "bayesprism",
n_cores = 12
)We can visualize the results as before:
omnideconv::plot_deconvolution(list("bayesprism" = deconvolution.results.bayesprism), "bar", "method", "Spectral")7. Deconvolution of the bulk data at a lower resolution
The considered single-cell breast cancer dataset includes cell-type
annotations at three levels of resolution: celltype_major,
celltype_minor, and celltype_subset, which
distinguish 9, 29, and 58 cell types respectively. The different
cell-type annotations can be accessed with:
single.cell.data$celltype_major # Major annotation
single.cell.data$celltype_minor # Minor annotation
single.cell.data$celltype_subset # Subset annotationThese additional annotations provide a cell-type classification at a
finer resolution. For instance, at the celltype_major
level, we only have the T cell population, while at the
celltype_minor level, we can distinguish between CD4+ and
CD8+ T cells. In the following, we will again perform deconvolution
analysis with DWLS but, this time, using the celltype_minor
information. We will subsample the dataset as before, this time
considering the second level of resolution for the cell types, and
extract the objects needed for deconvolution.
max_cells_per_celltype <- 200
sampled.metadata <- single.cell.data@meta.data %>%
rownames_to_column("barcode") %>%
group_by(celltype_minor) %>%
nest() %>%
mutate(n = map_dbl(data, nrow)) %>%
mutate(n = min(n, max_cells_per_celltype)) %>%
ungroup() %>%
mutate(samp = map2(data, n, sample_n)) %>%
select(-data) %>%
unnest(samp)
single.cell.data.sampled <- subset(single.cell.data, cells = sampled.metadata$barcode)| celltype_minor | number_cells |
|---|---|
| B cells Memory | 200 |
| B cells Naive | 200 |
| CAFs MSC iCAF-like | 200 |
| CAFs myCAF-like | 200 |
| Cancer Basal SC | 200 |
| Cancer Cycling | 200 |
| Cancer Her2 SC | 200 |
| Cancer LumA SC | 200 |
| Cancer LumB SC | 200 |
| Cycling PVL | 37 |
| Cycling T-cells | 200 |
| Cycling_Myeloid | 200 |
| DCs | 200 |
| Endothelial ACKR1 | 200 |
| Endothelial CXCL12 | 200 |
| Endothelial Lymphatic LYVE1 | 183 |
| Endothelial RGS5 | 200 |
| Luminal Progenitors | 200 |
| Macrophage | 200 |
| Mature Luminal | 200 |
| Monocyte | 200 |
| Myoepithelial | 200 |
| NK cells | 200 |
| NKT cells | 200 |
| Plasmablasts | 200 |
| PVL Differentiated | 200 |
| PVL Immature | 200 |
| T cells CD4+ | 200 |
| T cells CD8+ | 200 |
counts.matrix <- as.matrix(single.cell.data.sampled@assays$RNA@counts)
cell.type.annotations <- single.cell.data.sampled$celltype_minor
batch.ids <- single.cell.data.sampled$orig.ident
signature.matrix.dwls.minor <- omnideconv::build_model(
single_cell_object = counts.matrix,
cell_type_annotations = cell.type.annotations,
method = "dwls",
dwls_method = "mast_optimized"
)
deconvolution.results.dwls.minor <- deconvolute(
bulk_gene_expression = bulk.data,
model = signature.matrix.dwls.minor,
method = "dwls",
dwls_submethod = "DampenedWLS"
)We can visualize the results as before:
omnideconv::plot_deconvolution(list("dwls" = deconvolution.results.dwls.minor), "bar", "method", "Spectral")8. Comparison of cell fractions across conditions
We can consider as well the metadata provided with the original paper, which include patient’s data, cancer subtype information and treatment details. We can first harmonize the sample names, and then combine all this information with the deconvolution results in one dataframe.
patient.metadata <- read_excel("vignette_files/41588_2021_911_MOESM4_ESM.xlsx",
sheet = 1, skip = 3
) %>%
select(c(1, 4, 5, 11, 12))
colnames(patient.metadata) <- c("Sample", "Grade", "Cancer_type", "IHC_subtype", "Treatment")
patient.metadata$Sample <- gsub("-", "", patient.metadata$Sample) %>%
paste0("CID", .)
patients.results <- rownames_to_column(as.data.frame(deconvolution.results.dwls), "Sample") %>%
gather(key = "celltype", value = "cell_fraction", -Sample) %>%
left_join(patient.metadata, by = "Sample")Each condition has a different number of samples:
| IHC subtype | n |
|---|---|
| ER+ | 12 |
| HER2+ | 2 |
| HER2+/ER+ | 2 |
| TNBC | 8 |
We can visualize the estimated cell fractions across samples with a boxplot, and group the results by IHC subtype:
patients.results %>%
filter(!is.na(IHC_subtype)) %>%
ggplot(aes(x = IHC_subtype, y = cell_fraction, fill = IHC_subtype)) +
geom_boxplot(outlier.size = 0.5) +
facet_wrap(~celltype, scales = "free_y", ncol = 3) +
theme_bw() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "none"
) +
scale_fill_brewer(palette = "Set2") +
labs(
x = "IHC Subtype", y = "Cell Fraction",
title = "Cell fractions by IHC subtype (DWLS)"
)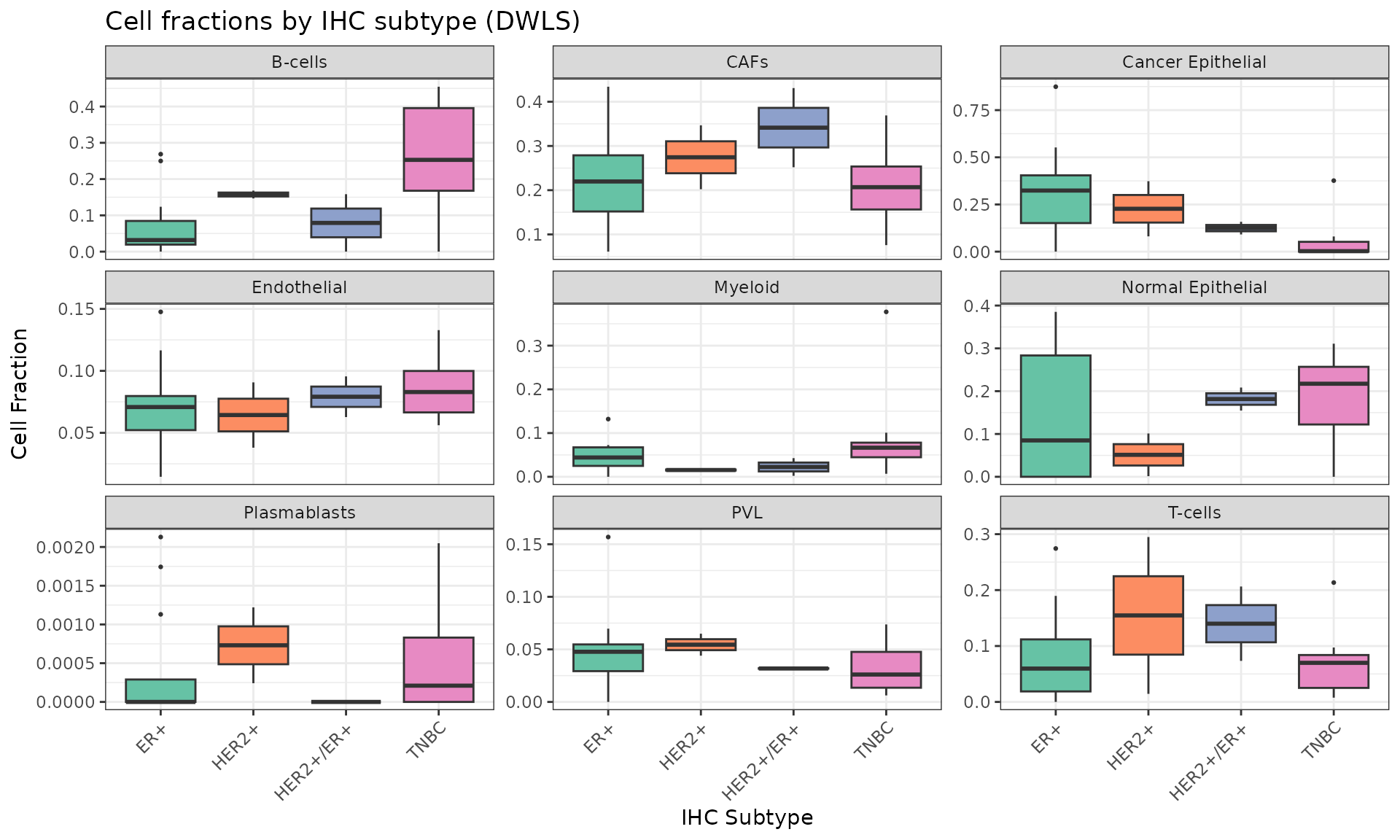
HER2-positive samples are enriched in T cells, while the ER-positive and triple negative breast cancer (TNBC) samples show an enrichment in cancer and normal epithelial cells, respectively. The patient metadata includes information about eventual treatments undergone by the patients. We can see that 5 out of the 24 patients were treated with Neoadjuvant and/or Paclitaxel, while the other 19 were untreated. We can therefore visualize the distribution of cell fractions across treated and untreated patients:
patients.results %>%
filter(!is.na(Treatment)) %>%
ggplot(aes(x = Treatment, y = cell_fraction, fill = Treatment)) +
geom_boxplot(outlier.size = 0.5) +
facet_wrap(~celltype, scales = "free_y", ncol = 3) +
theme_bw() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "none"
) +
scale_fill_brewer(palette = "Set1") +
labs(
x = "Treatment", y = "Cell Fraction",
title = "Cell fractions by treatment status (DWLS)"
)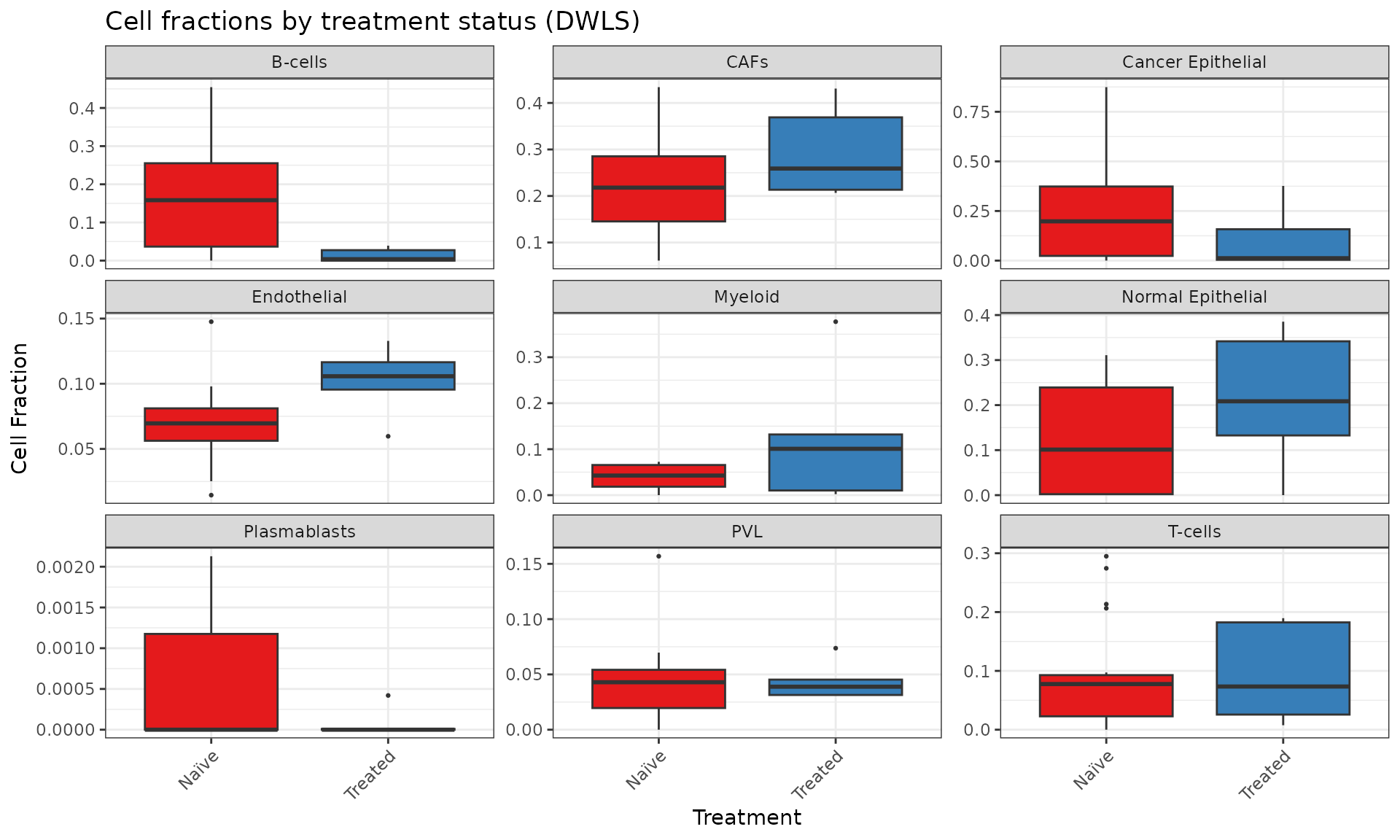
The major differences in this case are the enrichment of the CAFs and normal epithelial cells in the treated samples, as opposed to the cancer epithelial cells and B cells which have a higher median cell fraction in the untreated samples.
As mentioned before, with the lower level of annotations we can identify additional cell types, such as CD4+/CD8+ subtypes, Fibroblasts and cancer cells. We can therefore visualize the estimated cell fractions in a boxplot, focusing in particular on the CD4+ and CD8+ T-cell subtypes:
patients.results.minor %>%
filter(celltype %in% c("T cells CD4+", "T cells CD8+"), !is.na(IHC_subtype)) %>%
ggplot(aes(x = IHC_subtype, y = cell_fraction, fill = IHC_subtype)) +
geom_boxplot(outlier.size = 0.5) +
facet_wrap(~celltype, scales = "free_y") +
theme_bw() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "none"
) +
scale_fill_brewer(palette = "Set2") +
labs(
x = "IHC Subtype", y = "Cell Fraction",
title = "CD4+ and CD8+ T-cell fractions by IHC subtype"
)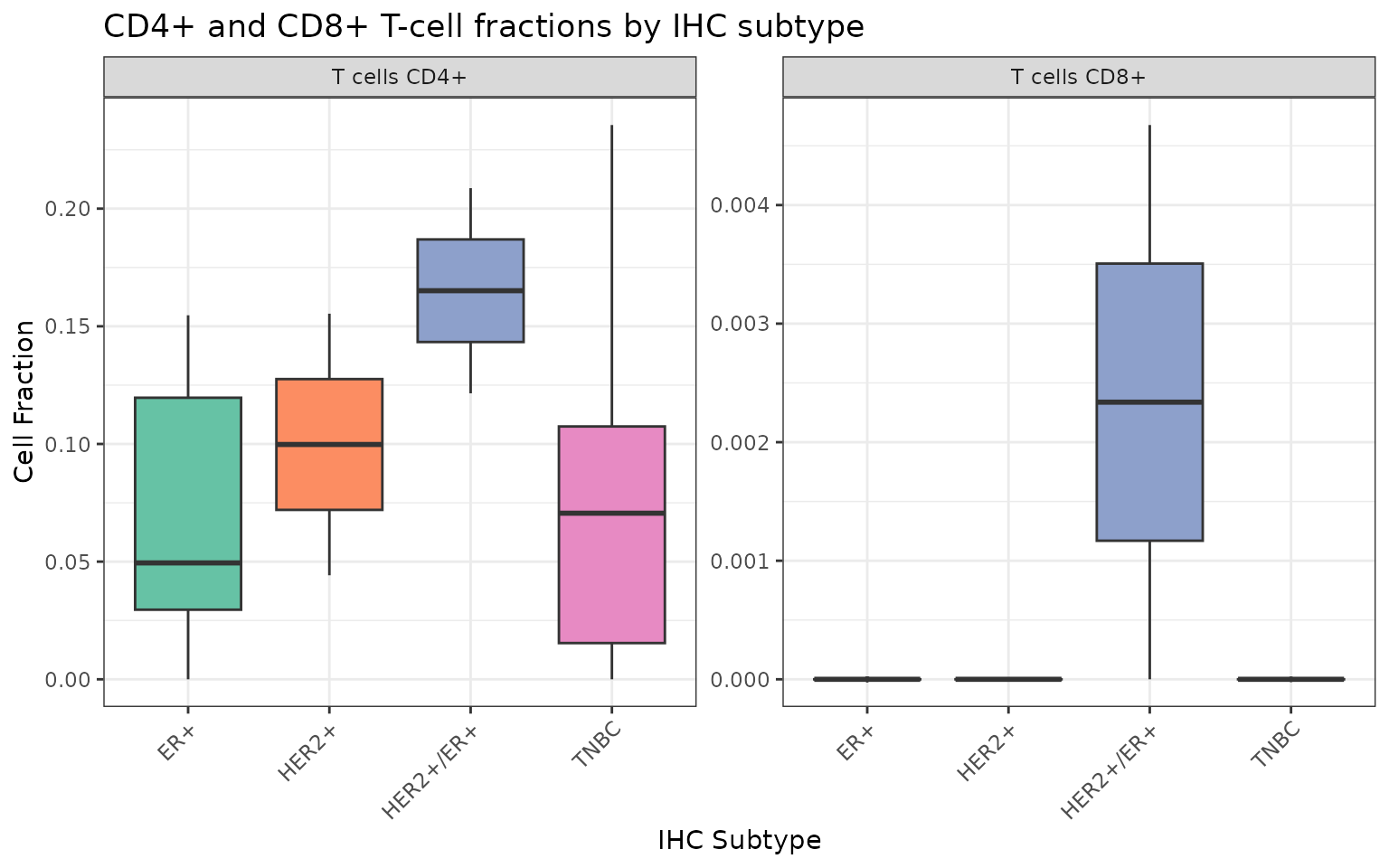
The estimates for the CD8+ T cells seem to be very low for all samples. On the other hand, in the HER2/ER positive samples the cell fractions estimated for the CD4+ T cells seem to be significantly higher than in the other subtypes.
Similarly, we can visualize the distribution of the estimates for the cancer associated fibroblasts (CAFs), respectively the myoblastic CAFs (myCAF) and the bone marrow derived inflammatory CAFs (MSC iCAF):
patients.results.minor %>%
filter(celltype %in% c("CAFs myCAF-like", "CAFs MSC iCAF-like"), !is.na(IHC_subtype)) %>%
ggplot(aes(x = IHC_subtype, y = cell_fraction, fill = IHC_subtype)) +
geom_boxplot(outlier.size = 0.5) +
facet_wrap(~celltype, scales = "free_y") +
theme_bw() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "none"
) +
scale_fill_brewer(palette = "Set2") +
labs(
x = "IHC Subtype", y = "Cell Fraction",
title = "CAF subtype fractions by IHC subtype"
)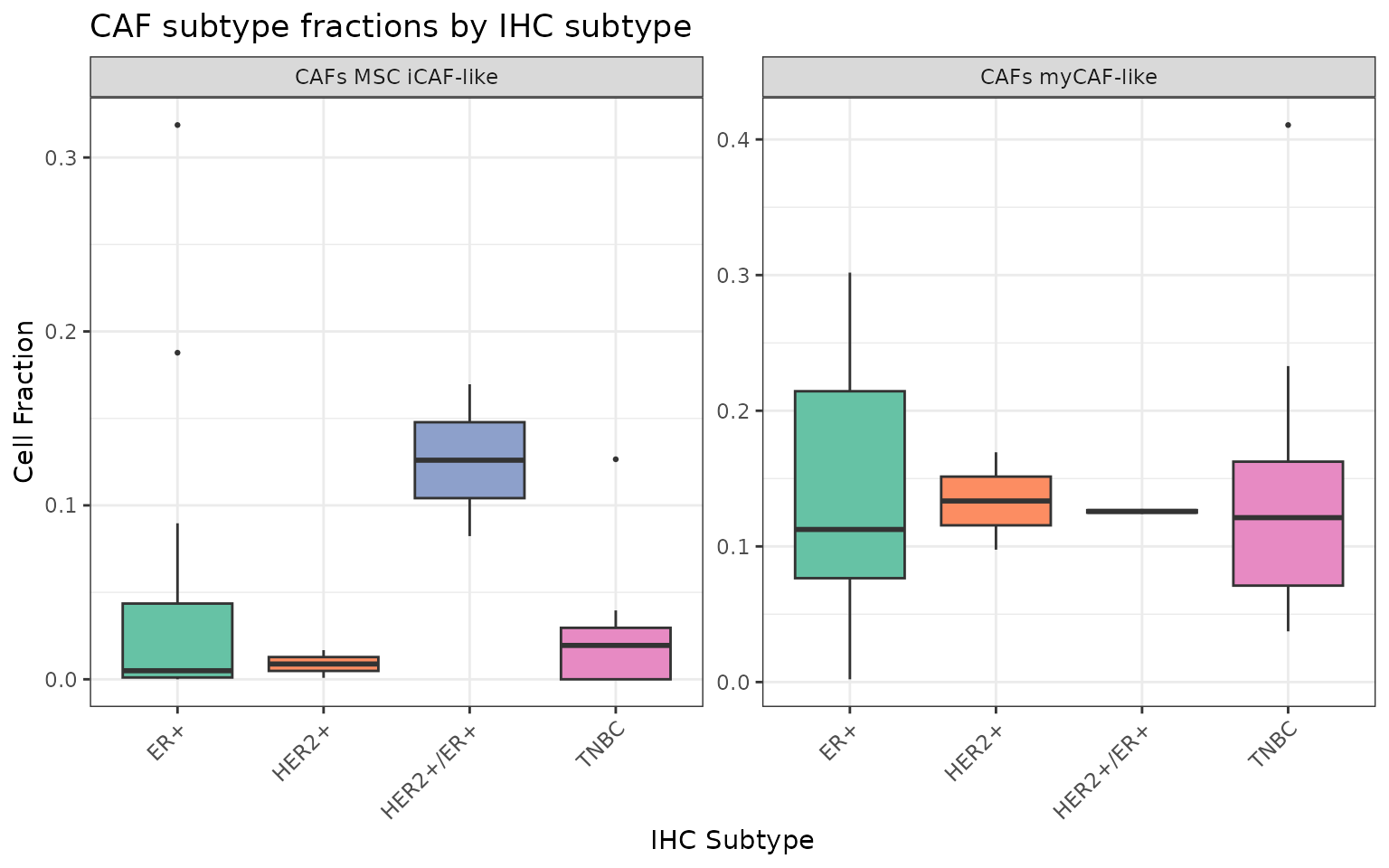
Here we can notice that, while the myoblastic CAFs have a comparable median value, the MSC CAFs have higher infiltration in the samples characterized as HER2+/ER+ subtype.
Finally, we can visualize the distribution of the different cancer cells molecular subtypes that were described by the authors:
patients.results.minor %>%
filter(grepl("^Cancer", celltype), !is.na(IHC_subtype)) %>%
ggplot(aes(x = IHC_subtype, y = cell_fraction, fill = IHC_subtype)) +
geom_boxplot(outlier.size = 0.5) +
facet_wrap(~celltype, scales = "free_y", ncol = 3) +
theme_bw() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "none"
) +
scale_fill_brewer(palette = "Set2") +
labs(
x = "IHC Subtype", y = "Cell Fraction",
title = "Cancer cell subtype fractions by IHC subtype"
)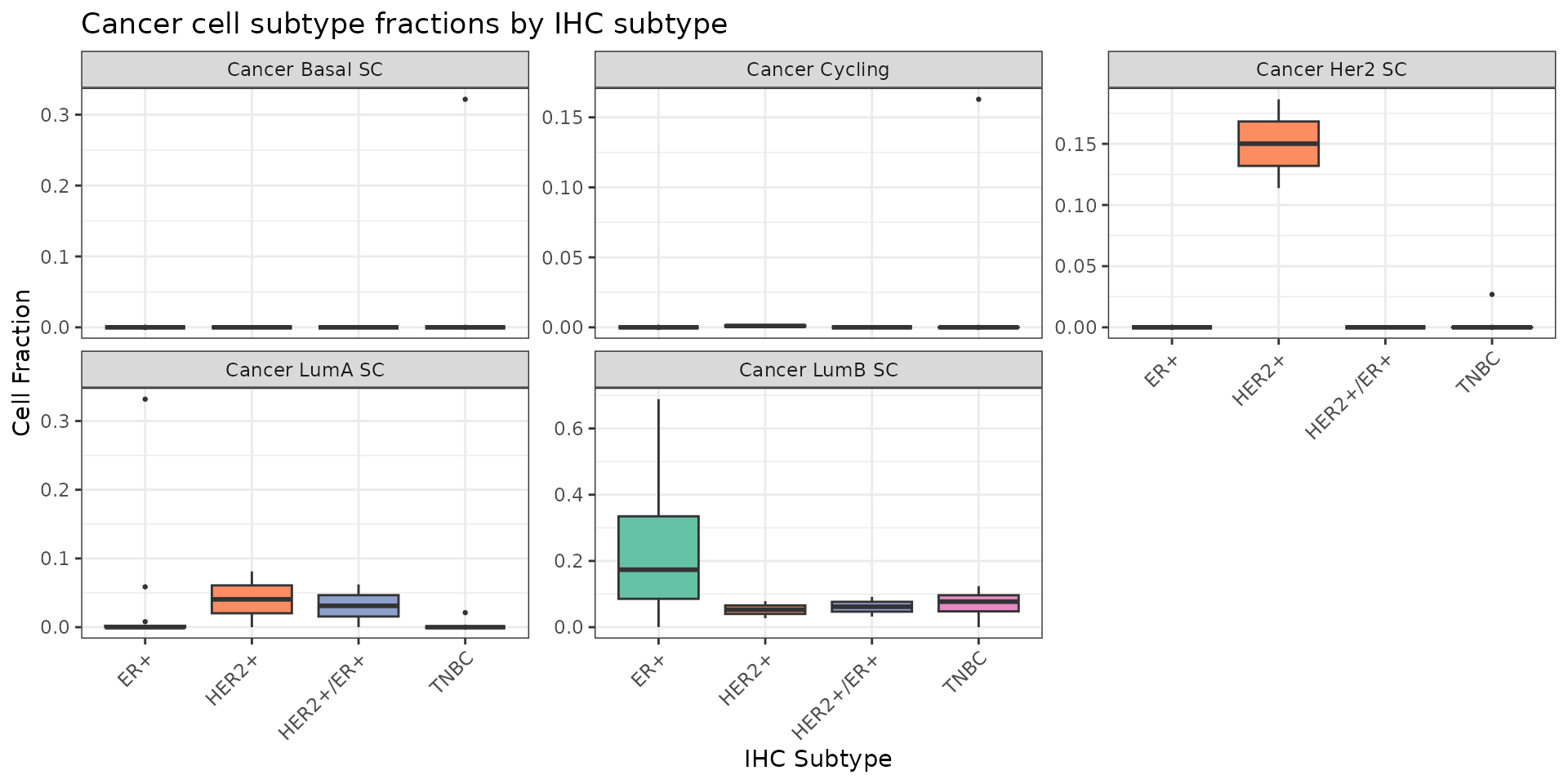
We can see that the HER2+ shows an overrepresentation of the cancer cells described as HER2-like by the authors. Similarly, the ER+ samples show higher fractions of Luminal B cancer cells. These findings on bulk RNA-seq are concordant with the subtypings that the authors described on the single cell RNA-seq (‘Results - scSubtype’).