Introduction
Methods like fluorescence activated cell sorting (FACS) or Immunohistochemistry (IHC)-staining have been used as a gold standard to estimate the immune cell content within a sample, however these methods are limited in their scalability and by the availability of good antibodies against the cell type markers. High throughput transcriptomic methods allow to get a transcriptional landscape in the sample with a relatively small amount of material that can be extremely limited in clinical settings (e.g. tumor biopsies), which led to high utility of methods like RNA-seq and microarrays to characterize patient tumor samples. However, RNA-seq does not provide a detailed information on a cellular composition of a sample, which then has to be inferred using computational techniques.
Conceptual differences between the methods
Such methods can, in general, be classified in two categories:
- Marker gene-based approaches and
- deconvolution-based approaches.
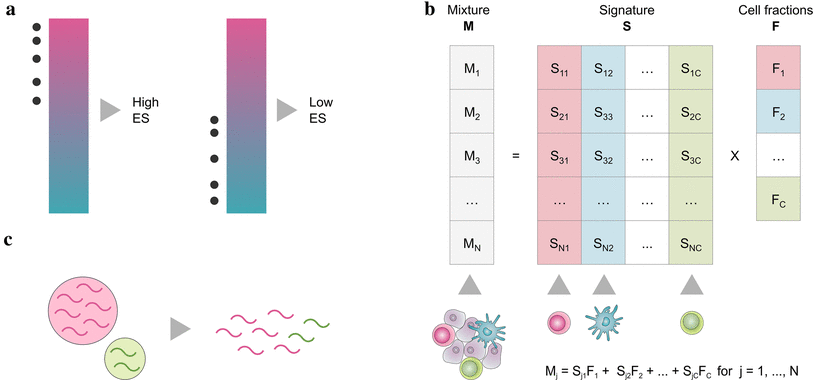
Marker gene based approaches (a) are based on a list of genes (signature), that are characteristic for a cell type. By looking at the expression values of signature genes, every cell type is quantified independently, either using the gene expression values directly (MCP-counter) or by performing a statistical test for enrichment of the signatures (xCell).
Deconvolution methods (b) formulate the problem as a system of equations that describe the gene expression of a sample as the weighted sum of the contributions of the different cell types. By solving the inverse problem, cell type fractions can be inferred given a signature matrix and the mixed gene expression. This can be accomplished using -Support Vector Regression (SVR) (CIBERSORT) constrained least square regression (quanTIseq, EPIC) or linear least square regression (TIMER).
For more information, check out the review by (Finotello and Trajanoski 2018).
Run the deconvolution
Input Data
The input data is a gene
sample gene expression matrix. In general values should
be
- TPM-normalized
- not log-transformed.
For xCell and MCP-counter this is not so important. xCell works on the ranks of the gene expression only and MCP-counter sums up the gene expression values.
Rownames are expected to be HGNC gene symbols. Instead of a matrix, immunedeconv also supports ExpressionSets (see below).
Run a method
This package gives you easy access to these methods. To run a method with default options, simply invoke
immunedeconv::deconvolute(gene_expression_matrix, method)where gene_expression_matrix is a matrix with genes in
rows and samples in columns. The rownames must be HGNC symbols and the
colnames must be sample names. The method can be one of
quantiseq
timer
cibersort
cibersort_abs
mcp_counter
xcell
epic
abis
consensus_tme
estimateThe ESTIMATE algorithm, which computes a score for the tumoral, immune and stromal components and the fraction of tumor purity of a sample, has been implemented.
immunedeconv::deconvolute_estimate(gene_expression_matrix)Deconvolution of mouse data
Imunedeconv has been extended to include methods aimed at the
deconvolution of mouse data. The format of the input
gene_expression_matrix is the same.
immunedeconv::deconvolute_mouse(gene_expression_matrix, method)The method can be one of
mmcp_counter
seqimmucc
dcq
baseIn addition, human-based methods can be used to deconvolute mouse data through the conversion of mouse gene names to the corresponding human orthologues
gene_expression_matrix <- immunedeconv::convert_human_mouse_genes(gene_expression_matrix, convert_to = "human")
immunedeconv::deconvolute(gene_expression_matrix, "quantiseq")Deconvolution using a custom signature
Finally, certain methods can be used with custom signatures, consisting of either a signature matrix or signature genes for the cell types of interest. Since the information used to deconvolute the bulk is user-provided, these functions can be used for different tissues and organisms. The functions may require different input data formats, related to the requirements of each method. Please refer to their documentation. The available methods are
cibersort norm/abs: deconvolute_cibersort_custom()
epic: deconvolute_epic_custom()
consensus_tme: deconvolute_consensus_tme_custom()
seqimmucc: deconvolute_seqimmucc_custom()Please note that using the
deconvolute_seqimmucc_custom() with a custom signature
matrix will automatically use the LLSR regression method. If you wish to
use the nuSVR regression method, use the
deconvolute_cibersort_custom() function.
Example
For this example, we use a dataset of four melanoma patients from (Racle et al. 2017).
res <- deconvolute(immunedeconv::dataset_racle$expr_mat, "quantiseq")## Warning: replacing previous import 'S4Arrays::makeNindexFromArrayViewport' by
## 'DelayedArray::makeNindexFromArrayViewport' when loading 'SummarizedExperiment'
knitr::kable(res, digits = 2)| cell_type | LAU125 | LAU355 | LAU1255 | LAU1314 |
|---|---|---|---|---|
| B cell | 0.02 | 0.43 | 0.02 | 0.49 |
| Macrophage M1 | 0.01 | 0.00 | 0.03 | 0.00 |
| Macrophage M2 | 0.00 | 0.00 | 0.00 | 0.01 |
| Monocyte | 0.18 | 0.00 | 0.00 | 0.00 |
| Neutrophil | 0.00 | 0.00 | 0.19 | 0.00 |
| NK cell | 0.05 | 0.00 | 0.00 | 0.00 |
| T cell CD4+ (non-regulatory) | 0.01 | 0.44 | 0.00 | 0.38 |
| T cell CD8+ | 0.00 | 0.03 | 0.09 | 0.05 |
| T cell regulatory (Tregs) | 0.02 | 0.10 | 0.06 | 0.06 |
| Myeloid dendritic cell | 0.00 | 0.00 | 0.00 | 0.00 |
| uncharacterized cell | 0.71 | 0.00 | 0.61 | 0.00 |
special case: CIBERSORT
CIBERSORT is only freely available for academic users and could not be directly included in this package. To use CIBERSORT with this package, you need to register on the cibersort website, obtain a license, and download the CIBERSORT source code.
The source code package contains two files, that are required:
CIBERSORT.R
LM22.txtNote the storage location of these files. When using
immunedeconv, you need to tell the package where it can
find those files:
library(immunedeconv)
set_cibersort_binary("/path/to/CIBERSORT.R")
set_cibersort_mat("/path/to/LM22.txt")Afterwards, you can call
deconvolute(your_mixture_matrix, "cibersort") # or 'cibersort_abs'as for any other method.
special case: TIMER and ConsensusTME
TIMER and ConsensusTME uses indication-specific reference profiles. Therefore, you must specify the tumor type when running TIMER or ConsensusTME:
deconvolute(your_mixture_matrix, "timer",
indications=c("SKCM", "SKCM", "BLCA"))indications needs to be a vector that specifies an
indication for each sample (=column) in the mixture matrix. The
indications supported by TIMER are
immunedeconv::timer_available_cancers## [1] "kich" "blca" "brca" "cesc" "gbm" "hnsc" "kirp" "lgg" "lihc" "luad"
## [11] "lusc" "prad" "sarc" "pcpg" "paad" "tgct" "ucec" "ov" "skcm" "dlbc"
## [21] "kirc" "acc" "meso" "thca" "uvm" "ucs" "thym" "esca" "stad" "read"
## [31] "coad" "chol"What the abbreviations stand for is documented on the TCGA wiki.
special case: seqImmuCC (mouse-based)
seqImmuCC is a method that can deconvolute using two regression approaches, SVR or LLSR. If the SVR approach is chosen, then the CIBERSORT script needs to be provided as described above.
Using ExpressionSets
The Bioconductor ExpressionSet is a convenient way to store a gene expression matrix with metadata for both samples and genes in a single object.
immunedeconv supports the use of an ExpressionSet
instead of a gene expression matrix. In that case, pData
requires a column that contains gene symbols. Which one needs to be
specified in the deconvolute() call:
deconvolute(my_expression_set, "quantiseq", column = "<column name>")Cell-type re-mapping
To provide consistently named results independent of the method, we defined a controlled vocabulary (CV) of cell-types and arranged them in a tree.
For each method, each cell-type is mapped to a node in the tree. If
you are curious, it’s all defined in this
excel sheet. This tree can be used to summarize scores along the
tree. For instance, quanTIseq provides scores for regulatory and
non-regulatory CD4+ T cells independently, but you are interested in the
fraction of overall CD4+ T cells. In that case you can use
map_result_to_celltypes to sum up the scores:
res <- deconvolute(immunedeconv::dataset_racle$expr_mat, "quantiseq") %>%
map_result_to_celltypes(c("T cell CD4+"), "quantiseq")
knitr::kable(res, digits = 2)| LAU125 | LAU355 | LAU1255 | LAU1314 | |
|---|---|---|---|---|
| T cell CD4+ | 0.03 | 0.54 | 0.06 | 0.44 |
The algorithm is explained in detail in the methods section of (Sturm et al. 2019).
Interpretation of scores
In general, cell-type scores allow for the comparison (1) between samples, (2) between cell-types or (3) both. Between-sample comparisons allow to make statements such as “In patient A, there are more CD8+ T cells than in patient B”. Between-cell-type comparisons allow to make statements such as “In a certain patient, there are more B cells than T cells”. For more information, see our Benchmark paper ((Sturm et al. 2019)).
Methods that allow between-sample comparisons
- MCP-counter
- xCell
- TIMER
- ConsensusTME
- ESTIMATE
- ABIS
- mMCP-counter (mouse based)
- BASE (mouse based)
Methods that allow both
- EPIC
- quanTIseq
- CIBERSORT abs. mode
- seqImmuCC (mouse based)
EPIC and quanTIseq are currently the only methods providing an absolute score, i.e. a score that can be interpreted as a cell fraction. These methods also provide an estimate for the amount of uncharacterized cells, i.e. cells for that no signature exists. This measure often corresponds to the fraction of cancer cells in the sample.
CIBERSORT abs., while allowing both between- and within-sample comparisons, generates a score in arbitrary units.
FAQs
Can I specify a custom signature matrix through immunedeconv?
Currentl, four methods (EPIC, CIBERSORT, ConsensusTME and seqImmuCC) allow users to provide their own signature matrix and/or gene set. Please note that the format of the signatures might vary across methods. In addition, since the embedded signatures were optimized for the methods, user-provided ones might have a worse performance. If you want to know more, please see #15.
I want to use a special feature of a method, but I cannot access it
through the deconvolute function.
You can access each method individually through the
deconvolute_xxx function. Through these functions you can
access all native features. See the function
reference for details.
If you believe that the feature is available across multiple methods
and should be added to the deconvolute interface, feel free
to open an issue or pull
request.